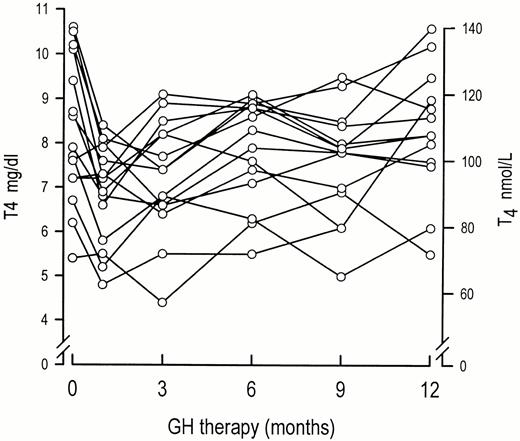
The present study demonstrates that peripheral T4 metabolism is subject to distinct dose-dependent regulation by GH. Although the unblinded and non-randomized design demands a cautious interpretation, it extends and supports the conclusion from earlier studies that the most important impact of GH administration on thyroid function is a stimulation of extrathyroidal T4 to T3 conversion (Msller et al., 1992; Sat0 et al., 1977; Rezvani et al., 1981; Grunfeld ef al., 1988; Jsrgensen et al., 1989; 1992; Pirazzoli et al., 1992). Conversely, our findings challenge the speculation that GH administration induces hypothyroidism. Such speculation derives mainly from observations of a decline in circulating T4 levels during GH therapy in GH-deficient children. In situations in which such a decline in T4 resulted in subnormal levels it is reasonable to assume that GH substitution had unmasked a state of incipient central hypothyroidism (Municchi et al., 1992). Our patients were adults, who had been on stable T4 supplementation prior to the study due to clinical hypothyroidism, which implies that the observed effects of GH on T4 metabolism occur at extrathyroidal sites and at the same time excludes a potentially confounding factor, which may be present, when studying patients with untreated subclinical central hypothyroidism. Nevertheless, our patients exhibited biochemical evidence of hypothroidism in terms of subnormal total and free T3 levels when not receiving GH therapy. This occurred in spite of total and free T4 levels well within the normal range. Similar observations have been made in a study of GH-deficient adults who did not receive T4 supplementation (Jsrgensen et al., 1989). Since T3 levels normalized following GH therapy in both studies it seems plausible that endogenous GH is a physiological regulator of peripheral iodothyronine metabolism and action. As an index of thyroid hormone action we included measurements of resting EE in our patients. A weak but significant positive correlation was recorded between FT3 and EE. Furthermore, resting EE was lower than predicted when the patients were studied without GH and this parameter was nor- malized by GH administration (Msller et al., 1993). These data may suggest that the GH induced increase (nor- malization) in T3 concentrations is associated with increased T3 action. On the other hand, it is possible that the calorigenic actions of GH may involve additional mechanisms, since an increase in EE has been observed following co-administration of GH in T3 treated obese patients (Bray et al., 1971). It is likely that the stimulation of peripheral T4 to T3 conversion contributes to the composite effects of GH treatment in both normal and GH-deficient patients. Apart from the increase in EE, the changes in iodothyronine metabolism could also be causally linked to other clinical effects such as the increase in heart rate and improvements in lipoprotein patterns and psychological well-being. On the other hand, the well known GH induced effects on bone and muscle mass, exercise capacity and muscle strength indicate that GH actions are mediated by several additional mechanisms. How GH treatment induces its effects on peripheral T4 metabolism remains unknown. During catabolic states a decreased peripheral T4 to T3 deiodination is an unequivo- cal finding (Cavalieri & Rapoport, 1977). The fact that such conditions (e.g. fasting, chronic illness, convalescence after surgery) are accompanied by elevated GH levels seems to suggest that the effects observed during GH therapy are not directly GH mediated but may involve additional factors such as metabolic factors and IGF-I generation. The introduction of biosynthetic IGF-I for in-vivo use in humans may provide a feasible means of further elucida- tion of these issues. We were surprised by the pronounced GH-induced suppression of circadian TSH levels. Notably, this effect was recorded in patients with substituted central hypo- thyroidism. The physiological significance is difficult to assess and complicated by reports from other studies suggesting that circulating TSH in such patients may possess reduced biological activity (Faglia et al., 1979). We also observed a pronounced suppression of TSH in the two patients without clinical evidence of hypothyroidism. Suppression of TSH in single measurements or a blunted TSH response to TRH during GH administration have also previously been reported (Root et ul., 1970; 1973; Lippe et ul., 1975; Grunfeld et al., 1988; Jsrgensen et al., 1989). It is tempting to suggest that the decline in TSH during GH administration is secondary to increased T3 generation within the pituitary. Interestingly, we were able to detect a significant nocturnal increase in serum TSH levels in the patients with central hypothyroidism when they did not receive GH. This pattern is comparable to that reported in normal subjects (Weeke, 1973; Rose h Nisula, 1989). It has been suggested that a deficient nocturnal surge of TSH is a sensitive marker of central hypothyroidism in both children and adults (Caron et al., 1986; Rose et ul., 1990). Our findings indicate that such a screening test should be interpreted with caution, in patients on concomitant GH therapy.
In summary, GH administration in GH-deficient adults is associated with pronounced dose dependent actions on thyroid function in terms of a stimulation of peripheral T4 to T3 conversion. Since the circulating T3 levels were subnormal in these patients when not receiving GH replacement, despite T4 substitution in conventional doses and normal T4 levels, we speculate that the effects of GH on peripheral iodothyronine metabolism may be physiologically important.

































.gif "IP Gear")